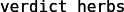
.prmtop
(wartość w polu Determine file type powinna się automatycznie
ustawić na AMBER7 Parm) i kliknij w przycisk
Load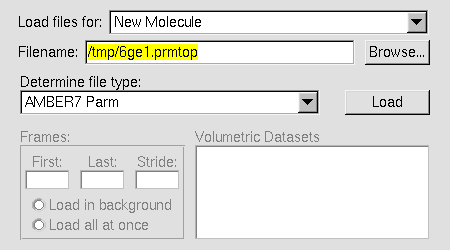
.nc (typ pliku powinien zostać rozpoznany jaki NetCDF
(Amber, MMTK)), kliknij w przycisk Load i zaobserwuj w
oknie VMD OpenGL Display wczytywane na bieżąco ramki

nucleicResNameNewCartoon
Wczytanie topologii i trajektorii:
from MDAnalysis.core.universe import Universe
topology = ... # ścieżka do pliku z topologią
trajectory = ... # ścieżka do pliku z trajektorią
u = Universe(topology, trajectory)Pole u.atoms zawiera wszystkie atomy widziane jako
obiekt klasy AtomGroup
Dowolny wybór atomów np. u.atoms[50:70]
również widziany jest jako obiekt klasy AtomGroup
Właściwość AtomGroup::residues zwraca obiekt klasy
ResidueGroup
Właściwość ResidueGroup::atoms zwraca wszystkie
atomy należące do reszty, zatem operacja
u.atoms[0:1].residues.atoms zwróci wszystkie atomy z
reszty, do której należy pierwszy atom
Właściwości ResidueGroup::resnames i
ResidueGroup::resids zwracają informację o nazwach i
numerach reszt opisywanych przez obiekt
Zarówno AtomGroup
jak i ResidueGroup
posiadają wiele użytecznych metod np.:
Universe lub AtomGroup
posiadają metodę select_atoms zwracającą
AtomGroupand, or,
notu.select_atoms('nucleic').residues.resnames pokaże nazwy
reszt kwasów nukleinowych (ale uwaga - reszty kwasów nukleinowych
rozpoznawane są wg listy predefiniowanych nazw)u.select_atoms("byres name C4'").residues.resnames (atom
C4’ występuje kwasach nukleinowych)u.select_atoms("not resname WAT and (not byres name C4')")Każda ramka trajektorii zawiera współrzędne wszystkich atomów w kolejnych krokach symulacji
W MDAnalysis każda iteracja po Universe::trajectory
wpływa na zmianę wszystkich współrzędnych, a więc i wyliczanych z nich
wartości
print(u.angles[0].value()) # wartość kąta w ramce 0
next(u.trajectory)
print(u.angles[0].value()) # wartość kąta w ramce 1Ogólny schemat analizy trajektorii wygląda tak:
features = []
for ts in u.trajectory:
frame = ts.frame # numer ramki
time = u.trajectory.time # punkt czasowy danej ramki
features.append(...) # wyznacz wartość w aktualnej ramce
... # agreguj wyniki cząstkoweW pakiecie MDAnalysis.analysis znajduje się wiele
interesujących funkcji np. do wyznaczania wartości RMSD:
r1 = u.select_atoms('resid 1')
A = r1.positions
u.trajectory[-1] # wybierz ostatnią ramkę
B = r1.positions
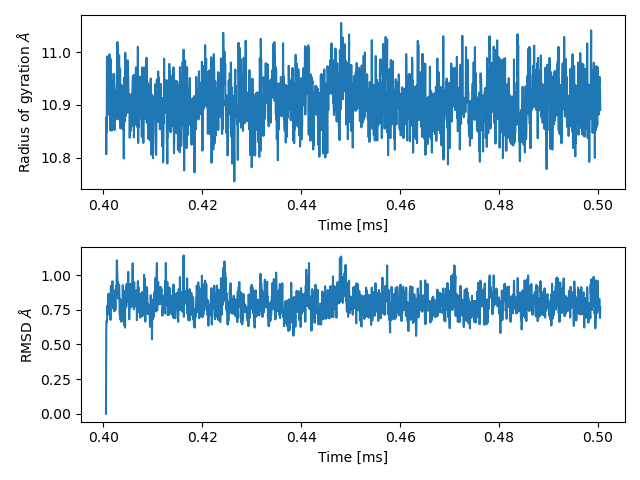
print(MDAnalysis.analysis.rms.rmsd(A, B))Przygotuj wykresy pokazujące wartość promienia bezwładności i
RMSD (względem współrzędnych z pierwszej ramki) dla struktury RNA w
każdej trajektorii (ocena 3.0)
Przyjrzyj się symulacji zapisanej jako pz31.nc.
Niektóre nukleotydy są wybrzuszone, więc podczas symulacji są bardziej
elastyczne.
Przeanalizuj ułożenie zasady azotowej dla nukleotydu o numerze 46.
Wykorzystaj wartość kąta torsyjnego \chi zdefiniowanego dla puryn jako
O4’-C1’-N9-C4. Przygotuj histogram wartości tego kąta w trajektorii
(ocena 4.0)

Uwaga! MDAnalysis pozwala wyliczyć wartość kąta torsyjnego opisanego
przez AtomGroup złożony z czterech atomów. Jednak bardzo
ważna jest kolejność atomów, a zaznaczając całą czwórkę jednym
wywołaniem select_atoms możemy otrzymać selekcję w dowolnej
kolejności. Dlatego w przypadku kątów torsyjnych, należy
AtomGroup stworzyć z połączenia czterech jednoatomowych
AtomGroup
W trajektorii 6GE1 mamy do czynienia z kwadrupleksem. To
czteroniciowy motyw, który najczęściej tworzą guaniny. Cztery zasady
azotowe tworzą tetradę dzięki niekanonicznym wiązaniom cis
Watson-Crick Hoogsteen. W idealnej tetradzie atomy tworzące cztery
zasady azotowe leżą na jednej płaszczyźnie, jednak w rzeczywistych
strukturach występują lokalne zaburzenia. Można je zmierzyć przy pomocy
parametru planarity deviation. W tym celu należy dla tetrady
wyznaczyć geometryczny środek atomów N9 oraz geometryczny środek atomów
C6. Wartością planarity deviation jest odległość między tymi
punktami.
Przygotuj wykres pokazujący wartość planarity deviation
zmieniającą się w czasie dla tetrady złożonej z reszt o numerach 2, 9,
16, 23 (ocena 5.0) 
Do wyznaczenia geometrycznego środka można użyć
AtomGroup::center_of_geometry(), które zwraca współrzędne
X, Y, Z punktu.