Pętla typu GNRA (gdzie N to dowolny nukleotyd, czyli
A, C, G lub U, a
R to puryna czyli A lub G) to
najczęstszy motyw strukturalny RNA
Cechą charakterystyczną motywu jest niekanoniczne wiązanie G-A
stabilizujące układ
Dwa pozostałe nukleotydy N i R tworzą
wiązania wodorowe z odległymi fragmentami jednoniciowymi lub
białkami
Utrzymują się w ten sposób przestrzenne interakcje wpływające na
całościowe zwinięcie się struktury
Biorą one udział w stabilizacji, szczególnie dla dużych struktur RNA
(1000+ nukleotydów)
Wizualizacja poniżej przedstawia strukturę drugorzędową dla 1ZIH
(tutaj pętla to GCAA):
Symulacje dynamiki
molekularnej
Symulacja dynamiki molekularnej to iteracyjne powtarzanie
rozwiązywania równań ruchu Newtona dla układu atomów (powiązanych
odpowiednimi ograniczeniami wynikającymi ze struktury i właściwości
własnych atomów)
Punktem początkowym jest struktura (współrzędne wszystkich jej
atomów), najczęściej umieszczona w wodzie z jonami celem
zobojętnienia
Układ posiada też temperaturę (czyli de facto wektory
prędkości dla każdego z atomów) i znajduje się pod określonym
ciśnieniem
Każdy krok symulacji odpowiada niewielkiej zmianie czasowej (często
1 fs, czyli 1e-15 s)
Symuluje się różnej długości trajektorie w zależności od problemu
badawczego
Dynamika molekularna pozwala też na:
sprawdzenie stabilności struktury po wprowadzeniu modyfikacji lub
mutacji,
analizę interakcji czwartorzędowych (np. przeprowadzenie tzw.
dokowania ligandu do miejsca aktywnego białka),
pełnoatomowe potwierdzenie eksperymentalnych wyników (np.
temperatura topnienia RNA/DNA to temperatura, w której 50% składu próbki
ulega denaturacji; przy pomocy symulacji można ten proces
prześledzić dokładniej),
przewidzenie struktury trzeciorzędowej na podstawie jej
sekwencji,
Początkowa struktura (współrzędne atomów) w formacie PDB (w
naszym przypadku: 1ZIH
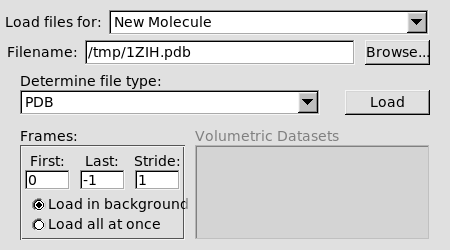
W okienku VMD Main wybieramy menu File → New
Molecule
Poprzez Browse wybieramy plik 1ZIH.pdb i
klikamy Load
Informacja o strukturze w formacie PSF:
W okienku VMD Main wybieramy menu Extension → Modeling
→ Automatic PSF Builder
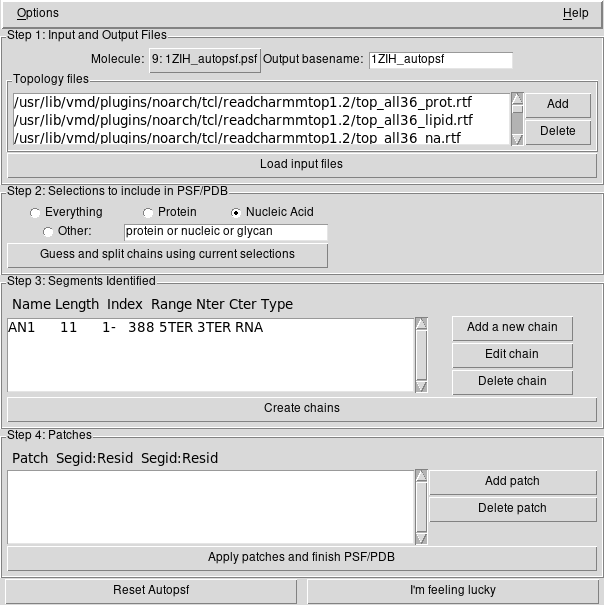
W okienku AutoPSF rozwijamy menu na górnym pasku
Options i zaznaczamy Add solvation box oraz Add
neutralizing ions
W sekcji Step 1: … klikamy Load input files
W sekcji Step 2: … zaznaczamy Nucleic Acid
W sekcji Step 2: … klikamy Guess and split …
W sekcji Step 3: … klikamy Create chains
Symulacja przy pomocy
AutoIMD (NAMD)
W okienku VMD Main wybieramy menu Extensions →
Simulations → AutoIMD (NAMD)
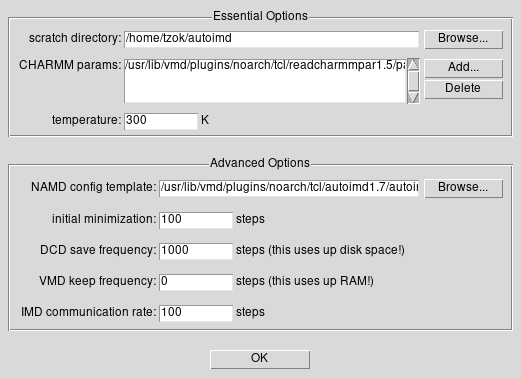
W okienku AutoIMD Controls wybieramy z menu na górnym
pasku Settings -> Simulation Parameters
Zmieniamy DCD save frequency na 1000 (jest to
częstotliwość zapisu “klatek” w trajektorii)
Zmieniamy IMD communication rate na 100 (jest to
częstotliwość aktualizowania VMD)
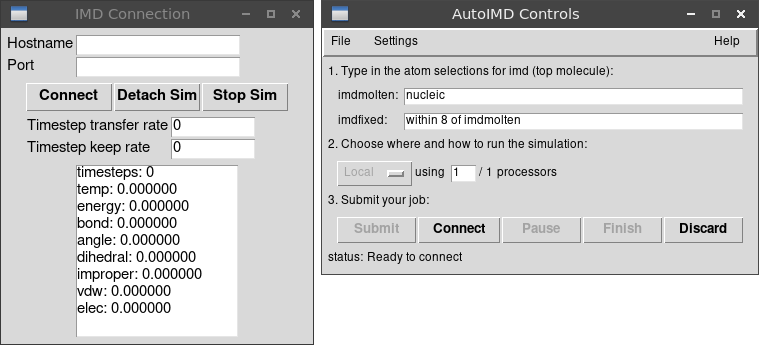
W okienku AutoIMD Controls wybieramy menu File →
Show Simulation Window
W okienku AutoIMD Controls w polu imdmolten
(ruchome atomy) wpisujemy nucleic i klikamy Submit,
ale jeszcze nie Connect
Zmieńmy sposób wizualizacji struktury RNA:
W okienki VMD Main mamy listę struktur i plików PSF,
dwukrotnie klikamy na litery D we wszystkich wierszach
oprócz AutoIMD Simulation. Dwukrotne kliknięcie zmieni
literę D na czerwoną i sprawi, że dane atomy nie będą
widoczne, zatem przestaniemy widzieć atomy tworzące wodę i jony
W okienku VMD Main wybieramy menu Graphics →
Representations
Ustawiamy Selected Molecule na AutoIMD
Simulation
Klikamy Create Rep
W polu Selected Atoms wpisujemy nucleic
Jako Drawing Method wybieramy HBonds
Jako Coloring Method wybieramy ColorID z
wartością 4
Możemy też zwiększyć Line Thickness do wartości 4
W głównym oknie VMD widzimy teraz tylko RNA oraz na żółto zaznaczone
wiązania wodorowe między zasadami
Gdy w okienku AutoIMD Controls klikniemy Connect
to uruchomi się symulacja, domyślnie etap
equilibration. Można oglądać efekty w głównym oknie VMD
oraz w logach w okienku IMD Connection
Etap ten kontynuuje się dopóki zachodzą jakieś znaczące zmiany, ale
w momencie gdy system zaczyna oscylować wokół tych samych stanów można
zatrzymać symulację klikając Pause (na potrzeby eksperymentu,
zatrzymajmy symulację po 10 tys. kroków = 20 ps czasu)
Tryb symulacji zmieniamy w okienku AutoIMD Controls w menu
Settings → Minimization Mode i uruchamiamy ciąg dalszy klikając
Pause. Minimalizację przeprowadza się dopóki symulacja nie
zatrzyma się w optimum lokalnym (na potrzeby eksperymentu, zatrzymajmy
symulację po 40 tys. krokach)
Analiza przebiegu symulacji
Do wykonania kroków poniżej sugeruję zacząć z
czystą sesją VMD:
W okienku VMD Main zaznaczamy wszystkie wiersze i wybieramy
z menu Molecule → Delete Molecule
Ewentualnie można wyłączyć i włączyć ponownie VMD :)
Namd generuje zestaw plików wyjściowych, a plugin AutoIMD
domyślnie umieszcza je w $HOME/autoimd. Nas
interesować będzie autoimd.log (tekstowy log z przebiegu
symulacji) oraz autoimd.dcd (binarnie zapisana trajektoria
= pozycje atomów zapisywane co pewną liczbę iteracji)
Aby otworzyć trajektorię w VMD:
Wybieramy z menu File → New Molecule
Upewnijmy się, że pole Load files for: ma wartość New
Molecule
Wybieramy plik autoimd.psf (Uwaga!
Lepiej wybrać plik PSF niż PDB. Dla tego drugiego wczytanie trajektorii
też zadziała, ale wówczas VMD nie skorzysta z pełni informacji o
strukturze jaka jest tylko w PSF)
Po naciśnięciu Load okienko wczytywania powinno
zostać aktywne, a pole Load files for: zmieni wartość na
autoimd.psf
Wybieramy plik autoimd.dcd i klikamy Load
Aby przeanalizować plik logu w VMD:
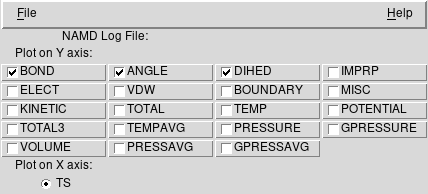
Wybieramy z menu Extensions → Analysis → NAMD Plot
W nowym okienku wybieramy File → Select NAMD Log File i
wskazujemy na plik autoimd.log
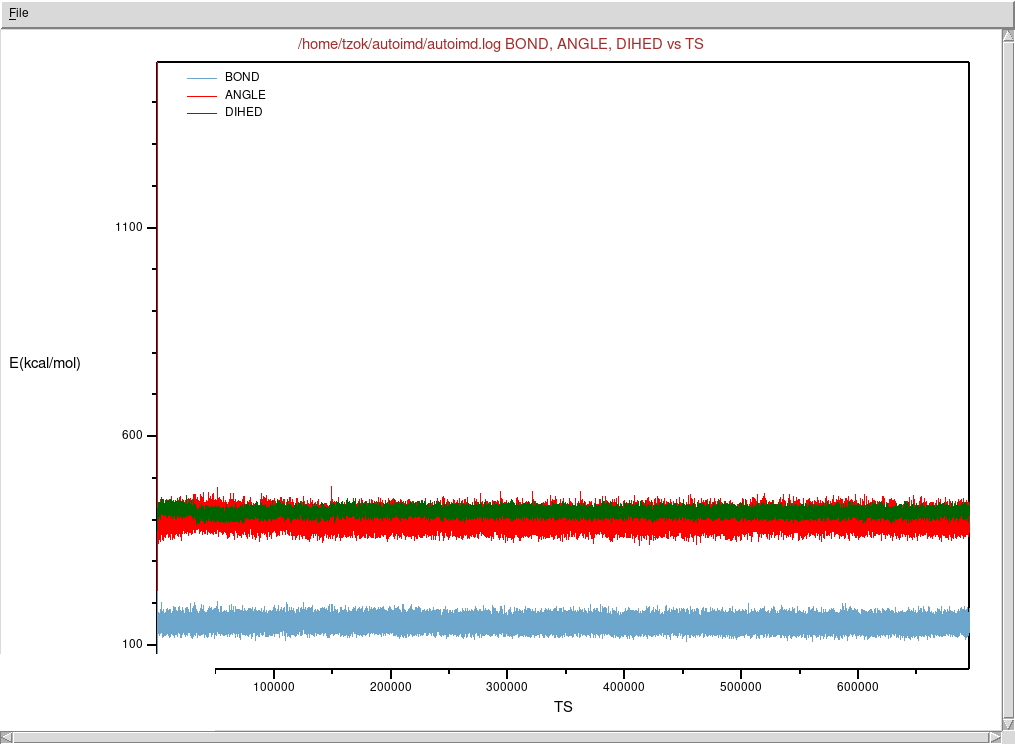
Zaznaczmy przykładowo BOND, ANGLE oraz
DIHED odpowiadające trzem komponentom funkcji energii w
modelu
Wybieramy z menu File → Plot Selected Data
Analiza gotowej, dłuższej
trajektorii
Pełniejszą i trójetapową symulację (minimalizacja, podgrzewanie,
swobodny ruch) możemy przeanalizować dzięki uprzejmości dr Joanny
Sarzyńskiej
Upewnijmy się, że Load files for: ma wartość New
Molecule
Wskazujemy na plik 1ZIH_js.psf (ponownie, nleży użyć
pliku PSF, a nie PDB) i klikamy Load
We wciąż otwartym oknie Molecule File Browser, z polem
Load files for: ustawionym na 1ZIH_js.psf
wskazujemy na plik min.dcd z pobranego archiwum i klikamy
Load
Powtaramy poprzedni krok dla heat.dcd i następnie
run1.dcd (w taki właśnie sposób “doczytuje” się kolejne
ramki z zapisanej trajektorii, VMD dokłada je na końcu tworząc spójny,
jednolity obraz symulacji)
Uruchom całą symulację (przycisk ▶ w oknie VMD Main),
możesz sterować suwakiem speed by
spowolnić wizualizację kolejnych kroków
Sprawdźmy przebieg zmian strukturalnych w czasie trwania
symulacji:
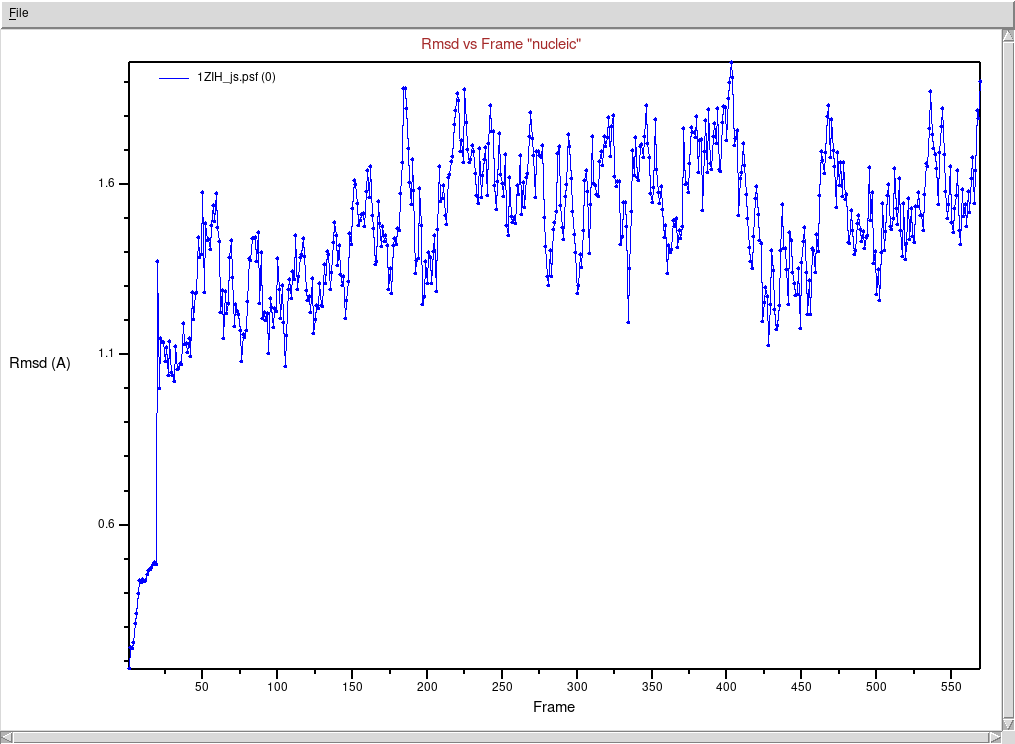
Wybieramy z menu głównego okna VMD Extension → Analysis → RMSD
Trajectory Tool
W polu edycyjnym w lewym górnym rogu (jest to zaznaczenie fragmentu,
którego ma dotyczyć analiza) wpisujemy nucleic
Klikamy najpierw przycisk ALIGN, następnie
RMSD
Wybieramy w menu File → Plot Data aby zobaczyć wykres
zmieniającego się RMSD w zależności od numeru ramki
(Uwaga! Różne etapy symulacji miały różny odstęp
czasowy pomiędzy zapisem współrzędnych do pliku trajektorii. Oznacza to,
że między sąsiednimi ramkami nie zawsze jest ta sama różnica w
czasie)
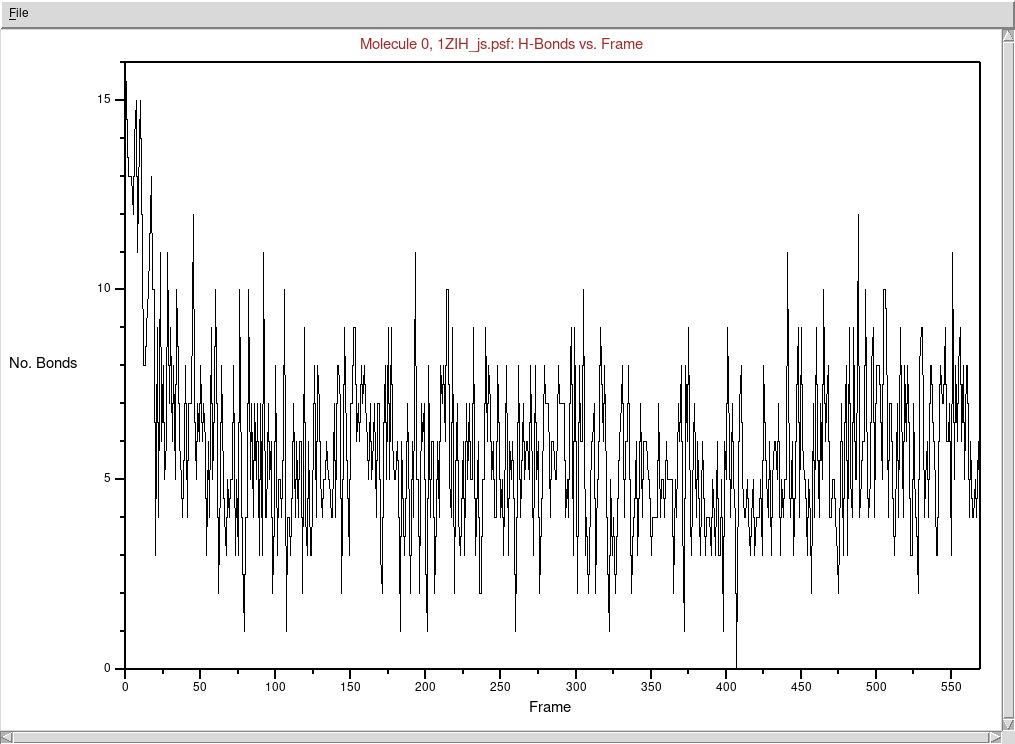
Sprawdźmy zmiany liczby wiązań wodorowych w czasie trwania
symulacji:
Wybieramy z menu głównego okna VMD Extensions → Analysis →
Hydrogen Bonds