Użyteczne linki:
{kind=link}
Wybór atomów w PyMOL-u
Do wyboru służy komenda: select. Mamy dwie podstawowe składnie:
select (selection)select name, (selection)
W przypadku pierwszym, tworzony jest domyślnie obiekt o nazwie sele. Kolejne wywołania nadpisują bez pytania to co było wcześniej w tym zapisane, dlatego w dłuższych sesjach warto nazywać wybrane grupy atomów samemu wykorzystując drugą składnię.
PyMOL posiada bardzo silny mechanizm wyboru atomów. Szczegóły można znaleźć na stronach Selection Algebra i Property Selectors w PyMOLWiki.
W powyższych wywołaniach select, wyrażenie (selection), może dotyczyć:
- typów atomów:
select symbol n - nazwanych atomów:
select name n1 - typów reszt:
select resn a5 - numerów reszt:
select resi 5 - nazw łańcuchów:
select chain a
W obrębie jednego wyrażenia, możemy podać więcej niż jeden selektor, np.:
- wybierz wszystkie atomy azotu i fosforu:
select symbol n+p - wybierz wszystkie atomy z reszt od 5 do 8:
select resi 5-8
Wreszcie PyMOL pozwala na łączenie wyrażeń zgodnie z zasadami teoriomnogościowymi, np.:
- wybierz wszystkie atomy azotu i fosforu, ale tylko wewnątrz reszt od 5 do 8:
select (symbol n+p) and (resi 5-8) - wybierz wszystkie atomy azotu w resztach od 10 do 12 i dodatkowo wszystkie atomy fosforu w resztach od 13 do 15:
select (symbol n and resi 10-12) or (symbol p and resi 13-15) - wybierz wszystkie atomy w łańcuchu A poza węglem:
select (chain a) and (not symbol c)
Są jeszcze operatory do zaznaczania wg odległości, wg wiązań i inne. Łącznie daje to naprawdę duże możliwości i opanowanie tej funkcjonalności jest kluczowe w wykorzystaniu całego potencjału PyMOL-a.
Podaj wyrażenia w języku selekcji PyMOL-a, które wybiorą poszczególne grupy atomów: (ograniczamy się do struktury: 1EHZ)
- Atomy węgla, ale tylko w szkielecie struktury (backbone), a nie w samych zasadach azotowych A, C, G i U
- Atomy fosforu w resztach o indeksie od 1 do 30 i atomy tlenu w pozostałych resztach
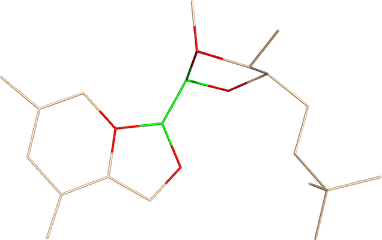
- Atomy tworzące wiązanie glikozydowe między pierścieniem rybozy i grupą (A, C, G lub U) oraz atomy z nimi związane. Przykładowo dla pojedynczej reszty jak na obrazku powinniśmy wybrać atomy zielone i dodać do grupy ich sąsiadów (atomy czerwone):
Różne sposoby reprezentacji
PyMOL jako narzędzie głównie do wizualizacji, posiada ogromne możliwości w generowaniu grafik wysokiej jakości. Oprócz szeregu przeróżnych typów wizualizacji możemy zmieniać jeszcze takie cechy jak przezroczystość czy światło. Warto zobaczyć przykłady na stronach All PyMOL Representations i w szczególności Cartoon na PyMOLWiki. Podgląd typów dostępnych dla kwasów nukleinowych znajduje się na stronie Examples of Nucleic Acids Cartoons.
Mając wczytaną strukturę, nowy sposób reprezentacji możemy dodać przy użyciu komendy: show representation, (selection). Jeśli chcemy dla pewnego zbioru atomów pozostawić tylko wybraną reprezentację, używamy komendy: show_as.
Jeśli (selection) jest puste, domyślnie dotyczy wszystkich atomów. W przeciwnym razie zawiera albo nazwany zbiór atomów, który już wybraliśmy wcześniej, albo wyrażenie ewaluowane jak przy normalnym zapytaniu select. Dzięki temu można np. wybrać interesujące nas reszty jako nazwany zbiór i następnie przy użyciu operatora and wizualizować pewne jego fragmenty w inny sposób:
show_as sticks, sele
show_as spheres, sele and symbol n
Najlepiej samemu sprawdzić jakie efekty daje wykorzystanie poniższych, najważniejszych reprezentacji:
- lines
- spheres
- mesh
- ribbon
- cartoon
- sticks
- dots
- surface
- everything
Przykładowo:
show_as everything
Domyślnie PyMOL pobiera z PDB (lub sam wyznacza jeśli w PDB brak informacji) dane dotyczące struktury drugorzędowej i automatycznie wizualizuje odpowiednio dane fragmenty struktury. Można jednak wymusić inne wyświetlanie przy pomocy komendy: cartoon type, (selection). Pole type może przyjmować następujące wartości:
- skip
- automatic
- loop
- rectangle
- oval
- tube
- arrow
- dumbbell
PyMOL pozwala nawet na precyzyjną kalibrację wymiarami odpowiednich typów reprezentacji. Np. dla tube można ustalić promień. Tu warto wspomnieć, że jest jeszcze dodatkowa reprezentacja zwana putty, która ustala ten promień na podstawie informacjo o b-factor pobranej z PDB.
Istnieją jeszcze zmienne binarne (przyjmujące wartości 0 lub 1), które przełączają inne cechy wizualizowanej struktury:
- cartoon_cylindrical_helices
- cartoon_fancy_helices
- cartoon_smooth_loops
Wypróbuj różne reprezentacji cząsteczki 2CFI. Podczas sprawdzania cartoon, sprawdź też komendy cartoon arrow, cartoon putty i pozostałe. A także poeksperymentuj ze zmiennymi np. set cartoon_smooth_loops, 1
Ciekawostka, grafiki typu old school :)
show_as cartoon
bg_color white
set ray_trace_mode, 2
set antialias, 2
ray
Wyświetlanie właściwości biochemicznych
Wartościowość
hide
select resi 1
zoom sele
orient sele
set valence, 1
bg_color white
ray
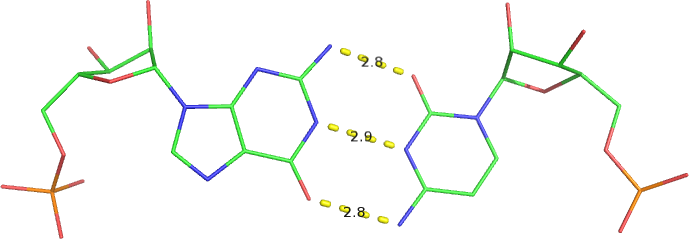
Wiązania wodorowe
select s1, resi 1
select s2, resi 72
select both, s1 or s2
hide
show_as lines, both
zoom both
orient both
distance hbonds, s1, s2, mode=2
bg_color white
ray
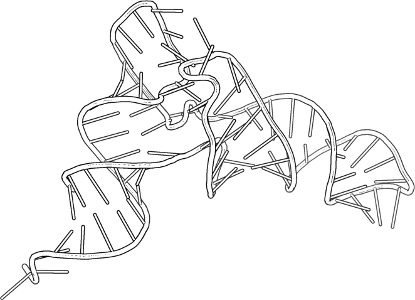
Poniżej przedstawiono G-quadruplex. Spróbuj możliwe najlepiej przedstawić tę strukturę (po lewej) przy pomocy metod jakie opisano powyżej. Struktura PDB to 2HY9. Docelowa grafika powinna zawierać taką pojedynczą "warstwę" jak na obrazku poniżej, pokazywać wiązania wodorowe, wartościowość oraz symbole atomów innych niż węgiel. Proszę w odpowiedzi na to zadanie przesłać nie tylko obrazek, ale też skrypt, najlepiej z komentarzami (w PyMOL-u robi się je jak w Pythonie, przez znak #). Tak, żebym mógł wykonać tylko pymol skrypt.pml i po chwili otrzymam grafikę zgodną z opisem powyżej.
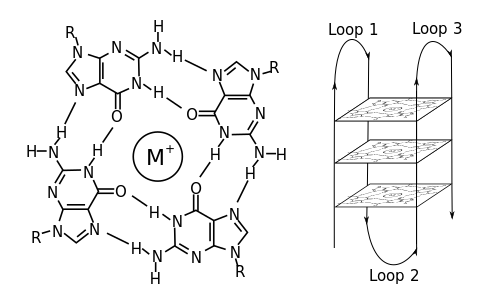
Uwaga
Po pierwsze, struktura 2HY9 nie zawiera centralnego jonu. Po drugie, przy automatycznym wykrywaniu wiązań wodorowych, PyMOL może wskazać na wielokrotne wiązania, mniej "eleganckie" niż na schematycznym rysunku (np. narysuje dwie linie idące od jednego wodoru - jedną do tlenu, drugą do azotu). To nie jest istotne, by te wiązania wyglądał identycznie jak na schemacie! Ważne jest by cztery wskazane zasady tworzyły G-quadruplex (każda guanina paruje się z dwiema sąsiednimi). Po trzecie, schemat pokazuje rybozę i szkielet fosforanowy jako po prostu R - na rysunku z PyMOL-a proszę albo zostawić rybozę/szkielet tak jak są, albo je schować zupełnie, a w każdym razie proszę nie tracić czasu na tym by koniecznie wyświetlić to jako R :)
Uwaga 2
Aby pokazać etykietę z symbolem atomu, należy wykorzystać składnię: label SELECTION, name[0], gdzie SELECTION oznacza wyrażenie, które wskaże odpowiednie atomy.
Integracja z Pythonem
PyMOL jest napisany w Pythonie i daje to dwustronne korzyści. Po pierwsze, umożliwia rozszerzenie funkcjonalności PyMOL-a poprzez własne skrypty. Po drugie, pozwala na wykorzystanie oferowanych przez funkcji "na zewnątrz" głównego programu, do własnych obliczeń.
Na Linuksie PyMOL integruje się zazwyczaj z systemowym Pythonem. Na Windowsie przygotowane paczki z PyMOL-em zawierają kopię Pythona, więc jeśli jest jakaś ustawiona systemowo wersja to ona nie będzie widzieć modułu pymol.
Domyślnie uruchomienie PyMOL-a wyświetla główne okienko. Aby tego uniknąć, należy rozpoczynać skrypty w następujący sposób:
__main__.pymol_argv = ['pymol','-qc']
import pymol
pymol.finish_launching()
# tutaj przychodzi nasz kod
Wszystkie komendy znane z PyMOL-a mają swoje odzwierciedlenie w Python-owym API, a dokładniej przez odpowiednie funkcje w module pymol.cmd. Przykładowo pymol.cmd.fetch('1ehz') ściągnie z Internetu strukturę 1EHZ. Ale nie jest to zupełnie jakbyśmy wykorzystywali bibliotekę bioinformatyczną, bo nie dostajemy w wyniku jakiegoś obiektu. Wywołanie takie sprawi po prostu, że struktura 1ehz pojawi się na liście obiektów w pamięci i choć nie mamy okienka, które to pokaże, można będzie wykonywać wszelkie inne komendy jak np. pymol.cmd.select('s1', '1ehz and resi 10'). O takiej specyficznej integracji z Pythonem trzeba pamiętać. Oczywiście można dostać się do danych zawartych w strukturze oraz niektóre komendy zwracają wynik w "regularny" sposób.
Jedną z takich komend jest fit, które nakłada na siebie dwa równoliczne zbiory atomów i oblicza ich RMSD. W PyMOL-u napisalibyśmy fit s1, s2, w Pythonie natomiast pymol.cmd.fit('s1', 's2'). I jak już podano wyżej, wywołanie takie zwraca wynik w sposób "regularny" tzn. jako liczbę rzeczywistą.
Tutaj pojawia się zysk jaki można osiągnąć z integracji między PyMOL-em i Pythonem. Przykładowo można wykorzystać biblioteki NumPy lub SciPy do dalszego przetworzenia wyników:
# coding=utf-8
# prolog wymagany przez PyMOL-a
import __main__
__main__.pymol_argv = ['pymol', '-qc']
import pymol
pymol.finish_launching()
# własne importy
import scipy.cluster.hierarchy
# PDB-id struktur, które nas interesują
IDS = ('1ehz', '1evv', '4tna', '4tra', '6tna')
if __name__ == '__main__':
# ściągnij każdą strukturę
for i in IDS:
pymol.cmd.fetch(i)
# przygotuj macierz porównań parami
count = len(IDS)
matrix = [[-1 for i in range(count)] for i in range(count)]
# porównaj parami każde dwie struktury (i wypisz od razu macierz)
print 'Matrix:'
for i in range(count):
print '[',
for j in range(i):
print '%0.2f' % matrix[i][j],
for j in range(i, count):
matrix[j][i] = matrix[i][j] = pymol.cmd.fit(IDS[i], IDS[j])
print '%0.2f' % matrix[i][j],
print ']'
# wykonaj klastrowanie wyników
# uznaj, że klastry oddzielone są o 3A
Z = scipy.cluster.hierarchy.average(matrix)
print scipy.cluster.hierarchy.fcluster(Z, 3, 'distance')
Wynik:
[ 0.00 1.11 0.95 6.26 7.09 ]
[ 1.11 0.00 1.12 1.21 1.19 ]
[ 0.95 1.12 0.00 1.83 1.84 ]
[ 6.26 1.21 1.83 0.00 7.68 ]
[ 7.09 1.19 1.84 7.68 0.00 ]
[2 1 1 3 4]
Struktury o tym samym indeksie należą do tego samego klastra. Przy poziomie 3 Angstremów, algorytm klastrowania połączył jedynie 1EVV i 4TNA, pozostałe struktury stanowią klastry jednoelementowe