Wstęp
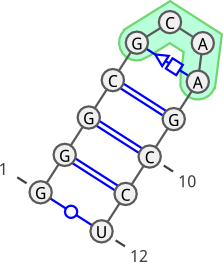
Pętle typu GNRA
- Pętla typu GNRA (gdzie N to dowolny nukleotyd, czyli A, C, G lub U, a R to puryna czyli A lub G) to najczęstszy motyw strukturalny RNA
- Cechą charakterystyczną wszystkich 8 możliwych układów jest niekanoniczne wiązanie G-A stabilizujące układ
- Dwa pozostałe nukleotydy N i R tworzą wiązania wodorowe z odległymi fragmentami jednoniciowymi lub białkami
- Utrzymują się w ten sposób przestrzenne interakcje wpływające na całościowe zwinięcie się struktury
- Biorą one udział w stabilizacji, szczególnie dla dużych struktur RNA (1000+ nukleotydów)
- Wizualizacja poniżej przedstawia strukturę drugorzędową dla 1ZIH (tutaj pętla to GCAA):
Symulacje dynamiki molekularnej
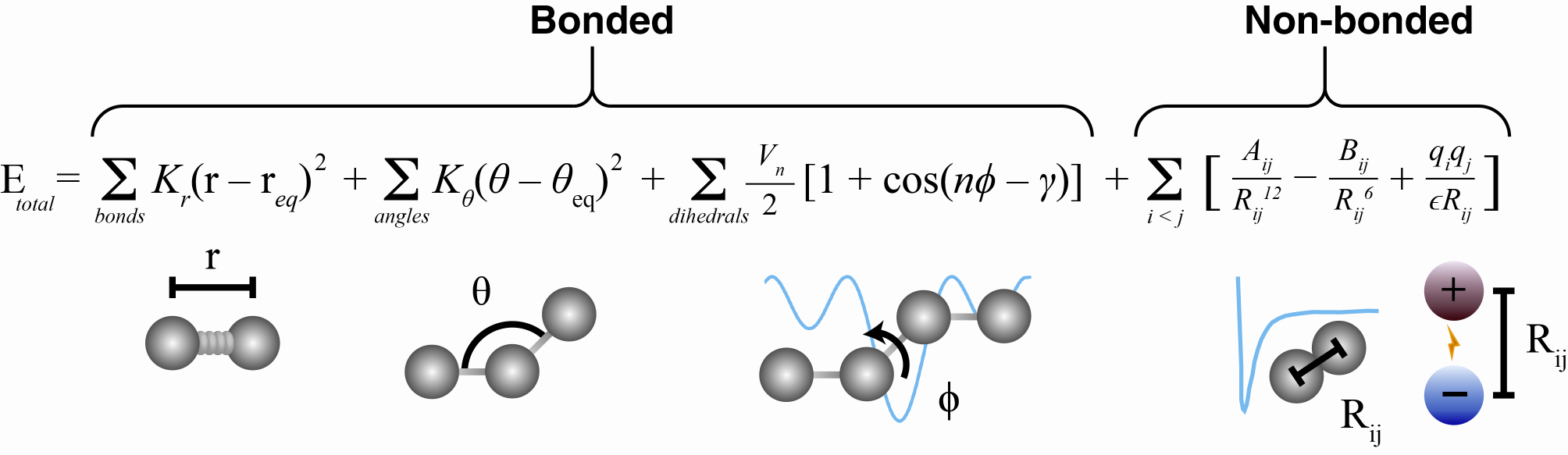
- Symulacja dynamiki molekularnej to iteracyjne powtarzanie rozwiązywania równań ruchu Newtona dla układu atomów (powiązanych odpowiednimi ograniczeniami wynikającymi ze struktury i właściwości własnych atomów)
- Punktem początkowym jest struktura (=współrzędne wszystkich jej atomów), najczęściej umieszczona w wodzie z jonami celem zobojętnienia
- Układ posiada też temperaturę (czyli de facto wektory prędkości dla każdego z atomów) i znajduje się pod określonym ciśnieniem
- Każdy krok symulacji odpowiada niewielkiej zmianie czasowej (często 1-2 fs [10-15 s])
- Symuluje się różnej długości trajektorie w zależności od problemu badawczego. Przykładowo, białka zwijają się w czasie rzędu od kilkudziesięciu mikrosekund do nawet kilku sekund. Przeciętny czas trwania symulacji jest rzędu milisekund [10-3 s], czyli kroków symulacji jest ~1012
- Dynamika molekularna pozwala też na:
- sprawdzenie stabilności struktury po wprowadzeniu modyfikacji lub mutacji,
- analizę interakcji czwartorzędowych (np. przeprowadzenie tzw. dokowania ligandu do miejsca aktywnego białka),
- pełnoatomowe potwierdzenie eksperymentalnych wyników (np. temperatura topnienia RNA/DNA to temperatura, w której 50% składu próbki ulega denaturacji; przy pomocy symulacji można ten proces prześledzić dokładniej),
- przewidzenie struktury trzeciorzędowej na podstawie jej sekwencji,
- i wiele innych...
- Poniższy film przedstawia symulację pętli GNRA, w której autorom udało się z formy w pełni rozwiniętej (unfolded) otrzymać końcową postać znaną z wielu innych badań strukturalnych: (referencja)
Przeprowadzenie własnej symulacji
Wymagane oprogramowanie
Pliki potrzebne do przeprowadzenia symulacji
- Początkowa struktura (współrzędne atomów) w formacie PDB (w naszym przypadku: 1ZIH: pętla GCAA, 10 modeli NMR)
- Pliki parametrów i topologii: (źródło)
- Informacja o strukturze w formacie PSF (w dalszej sekcji)
- Plik konfiguracyjny opisujący przebieg symulacji (w dalszej sekcji)
Wizualizacja w VMD
- Wczytaj strukturę do programu VMD (File → New molecule... → Browse...)
- Klik oraz rolka od myszy pozwalają na obrót oraz zbliżenie/oddalenie
- Domyślnie VMD wyświetla struktury w reprezentacji "Lines", zmień to następująco:
- Otwórz menu Graphics → Representations...
- Dla wybranej reprezentacji zmień Drawing Method na NewRibbons
- Dodaj reprezentację (przycisk Create Rep), ustaw Drawing Method na HBonds, Coloring Method na ColorID z wartością 1 red oraz Line Thickness równym 5
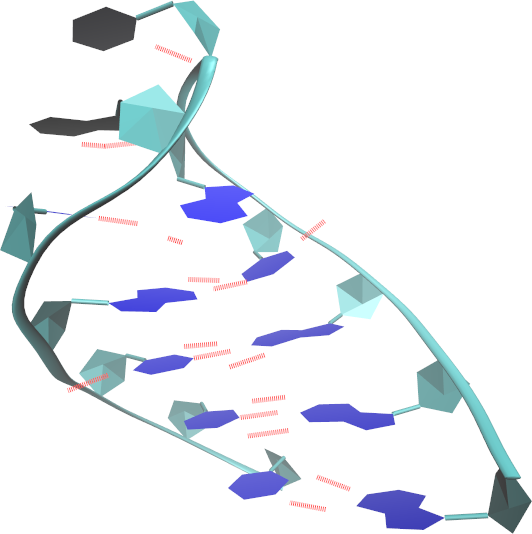
- Żeby przedstawić wszystkie 10 modeli na jednej wizualizacji:
- Wyłącz rysowanie wiązań wodorowych (dwukrotne kliknięcie na liście reprezentacji w HBonds)
- Kliknięciem wybierz reprezentację NewRibbons i w zakładce Trajectory, w polu Draw Multiple Frames wpisz: 1:10 (w ogólności od:do gdzie od i do to numery modeli/ramek w trajektorii)
- Żeby sprawdzić różnice pomiędzy modelami, wykorzystaj narzędzie Timeline:
- Wybierz menu Extensions → Analysis → Timeline
- W nowym okienku, wybierz menu Calculate → Calc. H-bonds... (z domyślnymi wartościami):
- na osi X pokazane są numery modeli
- na osi Y identyfikatory atomów
- czarny kolor oznacza brak wiązania wodorowego, biały natomiast jego obecność
- ta wizualizacja pozwala sprawdzić stabilność wiązań w warunkach eksperymentu NMR, ponieważ patrząc wierszami widać jak często wiązanie wystąpiło w różnych modelach

- Wybierz teraz menu Calculate → Calc. RMSD:
- na osi X ponownie pokazane są numery modeli
- na osi Y identyfikatory reszt
- pola wyświetlane są w skali szarości (patrz: legenda w lewym dolnym rogu)
- im jaśniejsze pole, tym bardziej dana reszta różni się od referencyjnej w pierwszym modelu (dlatego cała pierwsza kolumna jest czarna, RMSD wszędzie równe jest zero)
- na rys. w punkcie 4 powyżej możesz zauważyć dwie zasady azotowe w innym ułożeniu niż pozostałe 8; to jest właśnie reszta nr 6 (cytydyna) i obserwację te potwierdza wizualizacja z RMSD (dwa wyraźnie najjaśniejsze pola)
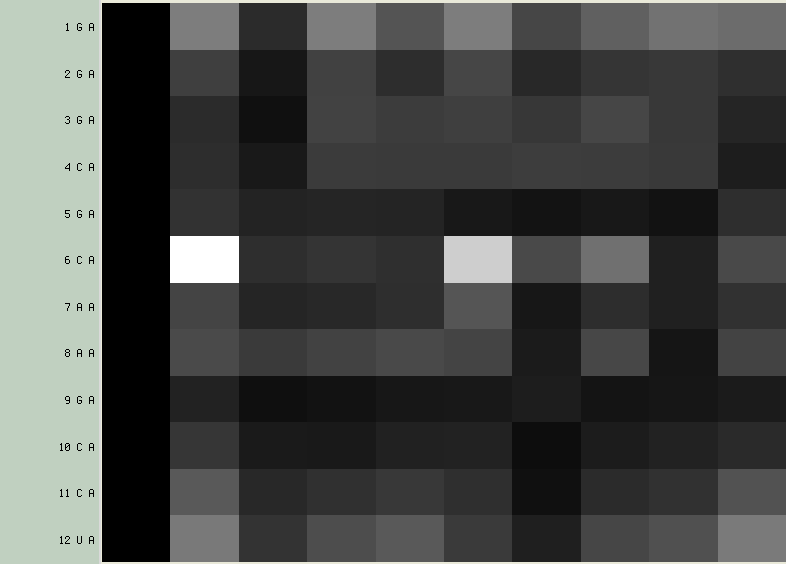
Przygotowanie pliku PSF (Protein Structure File)
- Plik PDB zawiera współrzędne atomów, natomiast plik PSF ma zapisane informacje o strukturze: masy atomów, wiązania, itp.
- Plik PSF buduje się w oparciu o strukturę PDB oraz topologię, można wykorzystać do tego narzędzie dostępne w VMD w menu Extensions → Modeling → Automatic PSF Builder:
- W menu Options zaznacz Add solvation box oraz Add neutralizing ions
- Upewnij się, że Molecule ustawione jest na 1ZIH.pdb
- Usuń domyślny plik z topologią i wskaż na ten ściągnięty wcześniej (plik: top_all36_na.rtf)
Uwaga! Zwróć uwagę na to, żeby był to plik z TOPologią, a nie PARametrami, czyli top_all36_na.rtf, a nie par_all36_na.prm - Kliknij w Load input files
- W kroku drugim zaznacz Nucleic Acid i kliknij w Guess and split chains using selections (później wskaż lokalizację pliku PDB gdy o to zapyta VMD)
- W kroku trzecim wybierz Create chains; wtyczka AutoPSF właśnie utworzyla nowy plik PDB (z przemianowanymi resztami, z atomami wody oraz z jonami Na+ i Cl-), a także plik PSF opisujący jego strukturę wewnętrzną
- Struktura umieszczona w wodzie z jonami jest automatycznie wczytana przez VMD, spróbuj zwizualizować strukturę ustawiając trzy aktywne reprezentacje w Graphical Representations:
- Style = NewCartoon, Color = ColorID 0, Selection = nucleic
- Style = VDW, Color = Name, Selection = ion
- Style = Licorice, Color = Name, Selection = water, Bond Radius = 0.1

Symulacja z wykorzystaniem periodycznych warunków brzegowych
- Ściągnij przedstawiony niżej szablon pliku konfiguracyjnego dla NAMD: (korzystając z linku poniżej
GetCode). Nazwij go np. namd.conf
- #############################################################
- ## JOB DESCRIPTION ##
- #############################################################
- # Minimization and Equilibration of
- # 1ZIH in a Water Box
- #############################################################
- ## ADJUSTABLE PARAMETERS ##
- #############################################################
- structure 1ZIH_autopsf.psf
- coordinates 1ZIH_autopsf.pdb
- set temperature 310
- set outputname 1ZIH_namd
- firsttimestep 0
- #############################################################
- ## SIMULATION PARAMETERS ##
- #############################################################
- # Input
- paraTypeCharmm on
- parameters par_all36_na.prm
- parameters par_water_ions.str
- temperature $temperature
- # Force-Field Parameters
- exclude scaled1-4
- 1-4scaling 1.0
- cutoff 12.0
- switching on
- switchdist 10.0
- pairlistdist 14.0
- # Integrator Parameters
- timestep 2.0 ;# 2fs/step
- rigidBonds all ;# needed for 2fs steps
- nonbondedFreq 1
- fullElectFrequency 2
- stepspercycle 10
- # Constant Temperature Control
- langevin on ;# do langevin dynamics
- langevinDamping 1 ;# damping coefficient (gamma) of 1/ps
- langevinTemp $temperature
- langevinHydrogen off ;# don't couple langevin bath to hydrogens
- # Periodic Boundary Conditions
- cellBasisVector1 48.0 0. 0.0
- cellBasisVector2 0.0 44.0 0.0
- cellBasisVector3 0.0 0 44.0
- cellOrigin 0.25 1.48 0.3
- wrapAll on
- # PME (for full-system periodic electrostatics)
- PME yes
- PMEGridSpacing 1.0
- #manual grid definition
- #PMEGridSizeX 45
- #PMEGridSizeY 45
- #PMEGridSizeZ 48
- # Constant Pressure Control (variable volume)
- useGroupPressure yes ;# needed for rigidBonds
- useFlexibleCell no
- useConstantArea no
- langevinPiston on
- langevinPistonTarget 1.01325 ;# in bar -> 1 atm
- langevinPistonPeriod 100.0
- langevinPistonDecay 50.0
- langevinPistonTemp $temperature
- # Output
- outputName $outputname
- restartfreq 500 ;# 500steps = every 1ps
- dcdfreq 250
- xstFreq 250
- outputEnergies 100
- outputPressure 100
- #############################################################
- ## EXTRA PARAMETERS ##
- #############################################################
- #############################################################
- ## EXECUTION SCRIPT ##
- #############################################################
- # Minimization
- minimize 100
- reinitvels $temperature
- run 2500 ;# 5ps
- Zwróć uwagę na zaznaczone linie:
- Przez 100 kroków odbywać się będzie minimalizacja energii układu, jest to obowiązkowy początek każdej symulacji dynamiki molekularnej
- Przez następne 2500 kroków, czyli przez 5 ps odbywać się będzie swobodna symulacja
- Umieść wskazane pliki w jednym katalogu wraz z namd.conf:
- 1ZIH_autopsf.pdb oraz 1ZIH_autopsf.psf (znajdują się one w katalogu VMD, czyli prawdopobnie:
C:\Program Files (x86)\University of Illinois\VMD) - par_all36_na.prm oraz par_water_ions.str (do ściągnięcia były wcześniej, linki na początku strony)
- 1ZIH_autopsf.pdb oraz 1ZIH_autopsf.psf (znajdują się one w katalogu VMD, czyli prawdopobnie:
- Popraw wymiary i położenie cząsteczki wraz z wodą i jonami:
- W VMD otwórz menu Extensions → Tk Console
- Zaznacz wszystkie atomy i wyznacz min-max:
set sel [atomselect top all]
set m [measure minmax $sel]
foreach {j1 j2} $m {}
foreach {x2 y2 z2} $j2 {}
foreach {x1 y1 z1} $j1 {} - Wyznacz poniższą wartość i wpisz ją jako pierwsze pole wektora cellBasisVector1 (pozostałe pola zostaw równe zero):
expr $x2 - $x1
- Wyznacz poniższą wartość i wpisz ją jako drugie pole wektora cellBasisVector2 (pozostałe pola zostaw równe zero):
expr $y2 - $y1
- Wyznacz poniższą wartość i wpisz ją jako trzecie pole wektora cellBasisVector3 (pozostałe pola zostaw równe zero):
expr $z2 - $z1
- Wyznacz środek i wpisz wynik jako cellOrigin:
measure center $sel
- Przygotuj plik uruchomieniowy do przeprowadzenia symulacji:
"C:\Ścieżka Do NAMD\namd2.exe" namd.conf > simulation.log
Analiza przebiegu symulacji
- Namd generuje zestaw plików wyjściowych, nas interesować będzie simulation.log (tekstowy log z przebiegu symulacji) oraz 1ZIH_namd.dcd (binarnie zapisana trajektoria = pozycje atomów zapisywane co pewną liczbę iteracji)
- Aby otworzyć trajektorię w VMD:
- Wybierz z menu File → New Molecule...
- Upewnij się, że pole Load files for: ma wartość New Molecule
- Wybierz plik 1ZIH_autopsf.psf (Uwaga! Wybierz plik PSF, a nie PDB. Dla tego drugiego wczytanie trajektorii też zadziała, ale wówczas VMD nie skorzysta z pełni informacji o strukturze jaka jest tylko w PSF)
- Po naciśnięciu Load okienko wczytywania powinno zostać aktywne, ale pole Load files for zawierać teraz będzie nazwę 1ZIH_autopsf.psf
- Wybierz plik 1ZIH_namd.dcd i kliknij w Load
- Aby przeanalizować plik logu w VMD:
- Wybierz z menu Extensions → Analysis → NAMD Plot
- W nowym okienku wybierz File → Select NAMD Log File i wskaż na plik simulation.log
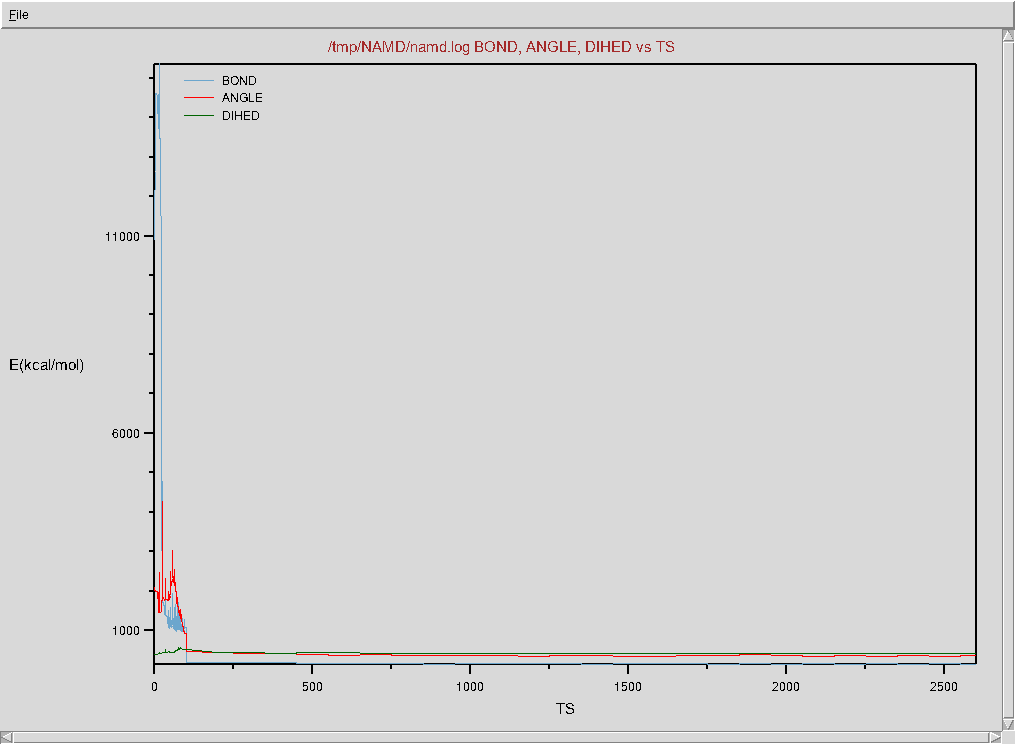
- Zaznacz przykładowo BOND, ANGLE oraz DIHED odpowiadające trzem komponentom funkcji energii w modelu
- Wybierz z menu File → Plot Selected Data, na wykresie wyraźnie widać moment przejścia symulacji z trybu minimalizacji energii (optymalizacji) do swobodnej symulacji cząsteczki
- Prześledź interakcję między resztami G-A (skrajne nukleotydy w pętli GNRA):
- Wybierz menu Graphics → Representations...
- Ustaw następującą wizualizację: (pogrubienie interesujących nas reszt oraz wyświetlenie atomów wody wokół nich)
- Style = Lines, Color = name, Selection = water and same resid as within 3.5 of (nucleic and resid 5 or resid 8)
- Style = Licorice, Color = name, Selection = nucleic and resid 5 or resid 8
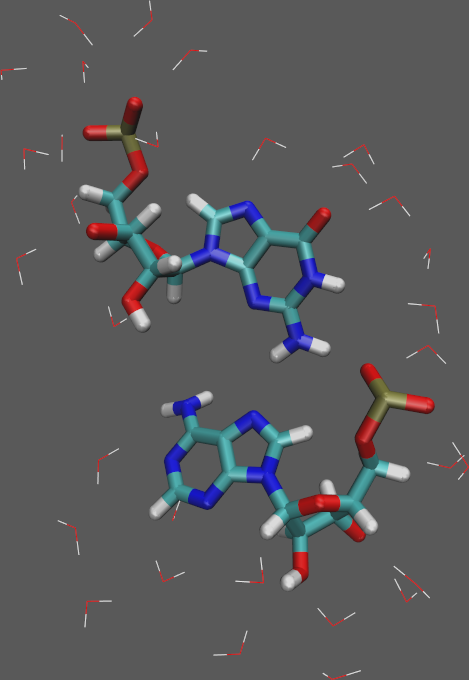
- W głównym oknie VMD przesuwaj po kolejnych zapisanych ramkach i obserwuj wzajemne ułożenie się reszt oraz cząsteczek wody
Analiza gotowej, dłuższej trajektorii
- Pełniejszą i trójetapową symulację (minimalizacja, podgrzewanie, swobodny ruch) możemy przeanalizować dzięki uprzejmości dr Sarzyńskiej:
- Aby wczytać pełną trajektorię:
- Pobierz i rozpakuj archiwum z wynikami oraz pliki PDB i PSF
- Wybierz w VMD File → New Molecule
- Upewnij się, że Load files for: ma wartość New Molecule
- Wskaż na plik 1ZIH_js.psf (ponownie, zwróć uwagę na to by użyć pliku PSF, a nie PDB) i kliknij w Load
- We wciąż otwartym oknie Molecule File Browser, z polem Load files for: ustawionym na 1ZIH_autopsf.psf wskaż na plik min.dcd z pobranego archiwum i kliknij w Load
- Powtórz poprzedni krok dla heat.dcd i następnie run1.dcd (w taki właśnie sposób "doczytuje" się kolejne ramki z zapisanej trajektorii, VMD dokłada je na końcu tworząc spójny, jednolity obraz symulacji)
- Uruchom całą symulację (przycisk "▶" w oknie VMD Main), możesz sterować suwakiem speed by spowolnić wizualizację kolejnych kroków
- Wykorzystaj narzędzie Timeline opisane wcześniej by zaobserwować otwieranie/zamykanie ostatniej pary zasad G1-U12
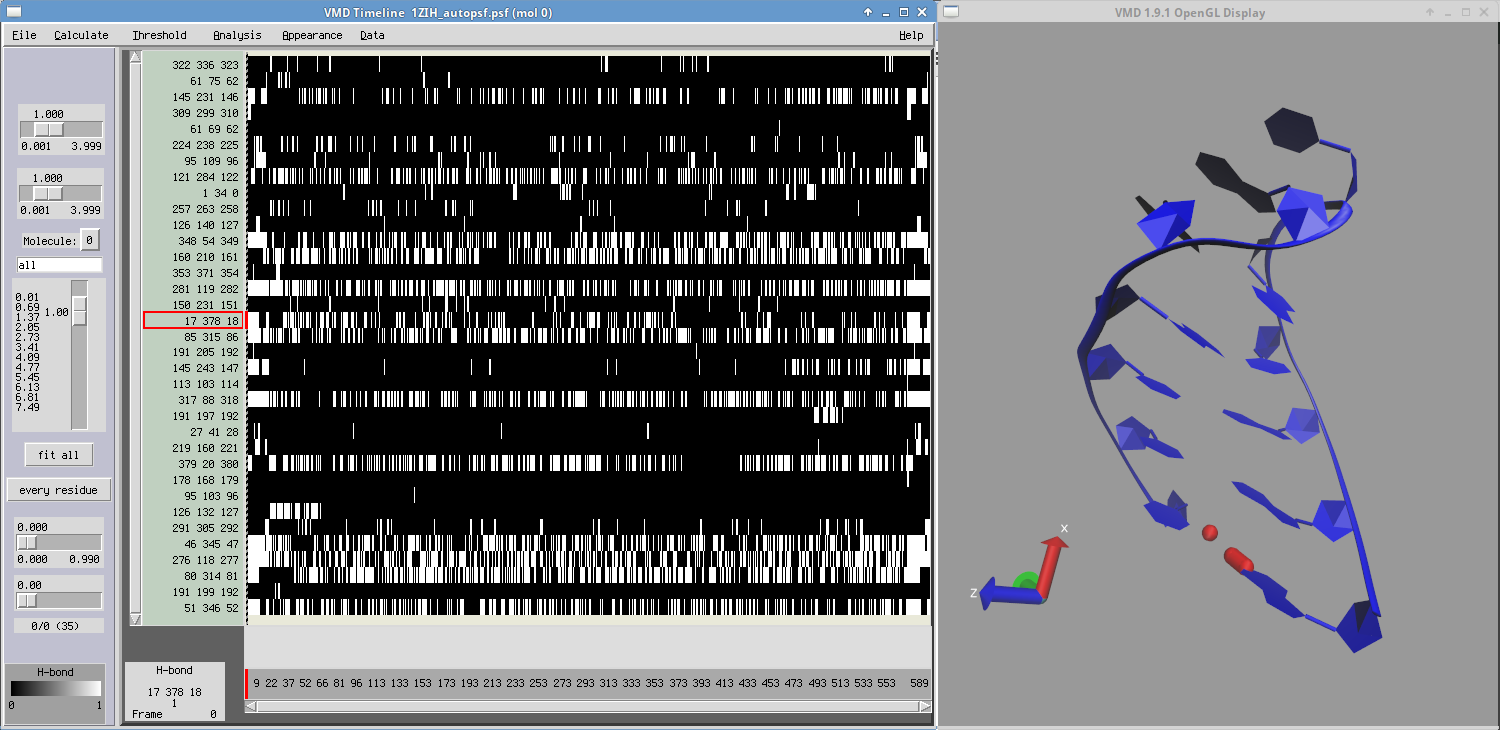
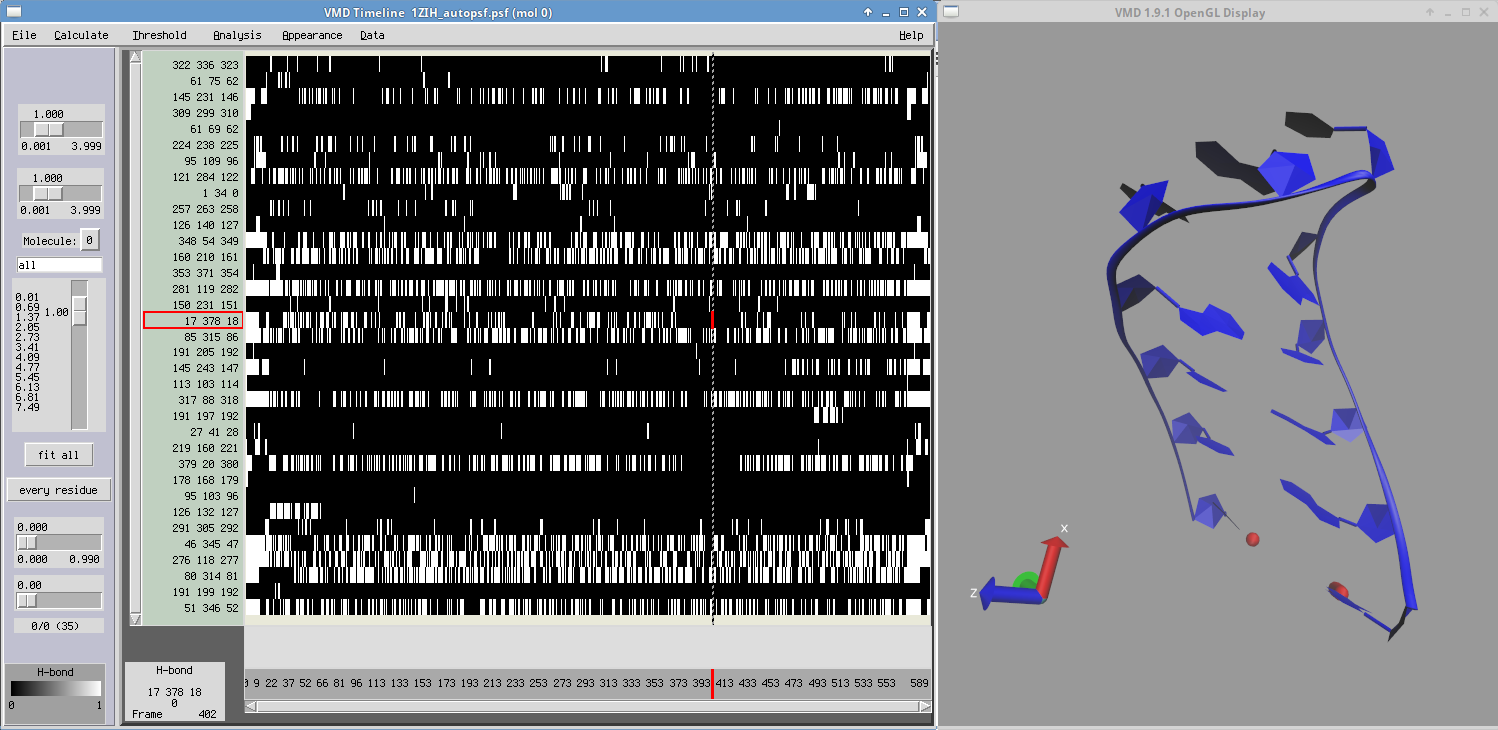
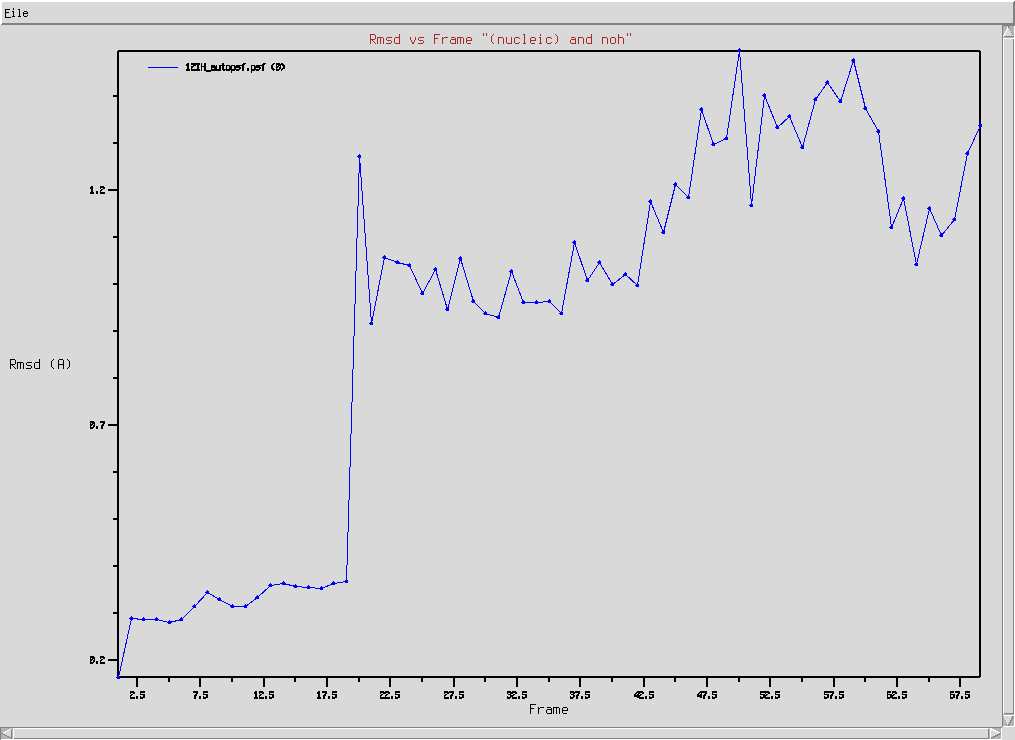
- Sprawdź przebieg zmian strukturalnych w czasie trwania symulacji:
- Wybierz z menu głównego okna VMD Extension → Analysis → RMSD Trajectory Tool
- W polu edycyjnym w lewym górnym rogu (jest to zaznaczenie fragmentu, którego ma dotyczyć analiza) wpisz nucleic
- Kliknij najpierw w przycisk ALIGN, następnie w RMSD
- Wybierz w menu File → Plot Data by zobaczyć wykres zmieniającego się RMSD w zależności od numeru ramki (Uwaga! Różne etapy symulacji miały różny odstęp czasowy pomiędzy zapisem współrzędnych do pliku trajektorii. Oznacza to, że między sąsiednimi ramkami nie zawsze jest ta sama różnica w czasie)